This is a user-friendly manual for biologists who are interested in performing quantitative trajectory inference with the Treefit package. If you are not familiar with the basic workflow of Treefit, then it would be a good idea to look at the introductory tutorial (vignette("treefit")) before using it.
1. Why “quantitative” trajectory inference?
Single-cell technologies are expected to help us discover a novel type of cells and revolutionize our understanding of the process of cell differentiation, and trajectory inference is one of the key computational challenges in single-cell transcriptomics. In recent years, many software packages have been developed and widely used to extract an underlying “tree” trajectory from single-cell RNA-seq data.
However, trajectory inference often suffers from the uncertainty due to the heterogeneity of individual cells and the high levels of technical noise in single-cell experiments. Hence, it was a fatal problem that there was no method to quantitatively assess the reliability of the estimated trajectory and to distinguish distinct cell types in an objective manner so far. Of course, many handy software packages are available for visualizing data and helping perform exploratory data analysis; however, we must stress that different visualization techniques often result in completely different pictures and the interpretation can be quite subjective and thus it is hard to reach a scientific truth solely by this approach. In order to facilitate scientific discoveries from single-cell gene expression data, we need a quantitative methodology.
This is why we developed Treefit - the first software for performing quantitative trajectory inference. Treefit can be used in conjunction with existing popular software packages such as Seurat and dynverse. In this tutorial, we demonstrate how to use Treefit together with Seurat by analyzing the single-cell RNA-seq dataset in Trapnell el al., 2014, Nature Biotechnology.
2. Trajectory inference using Treefit and Seurat
As the Treefit package is meant to be used with gene expression data (either raw counts or normalized values) that have been already quality controlled, it is a good idea to use Seurat or other popular toolkits designed for the quality control of single-cell gene expression data (e.g., filtering cells and normalizing the data) before the Treefit analysis.
2.1. Loading the dataset
We analyze the dataset provided in Trapnell el al., 2014, Nature Biotechnology. This dataset was originally acquired for the purpose of studying the process of differentiation of human myoblasts that can be reasonably explained by the linear model (i.e., non-branching, chain-like structure). However, as the authors discuss in detail in the original article, their trajectory inference software based on a minimum spanning tree (MST) helped detect contamination with another cell lineage which was not related to myogenesis and so revealed that the bifurcation tree model (i.e., Y-shaped tree structure) fits the data better than the linear model.
To use this dataset, we download and read the dataset provided in dynverse as follows (see https://zenodo.org/record/1443566 for details).
trapnell.path <- "myoblast-differentiation_trapnell.rds"
if (!file.exists(trapnell.path)) {
download.file("https://zenodo.org/record/1443566/files/real/gold/myoblast-differentiation_trapnell.rds?download=1",
trapnell.path,
mode="wb")
}
trapnell.dynverse <- readRDS(trapnell.path)2.2. Preprocessing with Seurat
From now, we demonstrate how to preprocess the above data with Seurat. In order to handle data in Seurat, we first need to create a Seurat object. When creating a Seurat object from dynverse data, we must be aware that the roles of the rows and columns are reversed and therefore we need to transpose the matrix. In dynverse data, the rows and columns correspond to cells and genes, respectively, and vice versa in Seurat objects.
Here we assume that we wish to perform the following preprocessing.
- Excluding each gene that is only expressed in less than 3 cells
- Removing each cell that only expresses less than 200 genes
This can be done by setting the designated parameters as follows.
trapnell <- Seurat::CreateSeuratObject(counts=t(trapnell.dynverse$count),
min.cells=3,
min.features=200)## Warning: Feature names cannot have underscores ('_'), replacing with dashes
## ('-')The “Feature names cannot have underscores …” warning message can be ignored. It’s caused by difference between dynverse and Seurat. Dynverse uses underscores for gene names but Seurat doesn’t allow it. Seurat replaces underscores with dashes automatically with the warning message.
2.3. Visualization with Seurat
Although pictures may not always describe reality, we usually visualize data before starting detailed analysis. Here we create a scatter plot by principal component analysis (PCA). Below are a few lines of code to do this and the result is shown in Figure 1.
trapnell <- Seurat::FindVariableFeatures(trapnell, verbose=FALSE)
trapnell <- Seurat::ScaleData(trapnell, verbose=FALSE)
trapnell <- Seurat::RunPCA(trapnell, verbose=FALSE)
plot(Seurat::Embeddings(trapnell))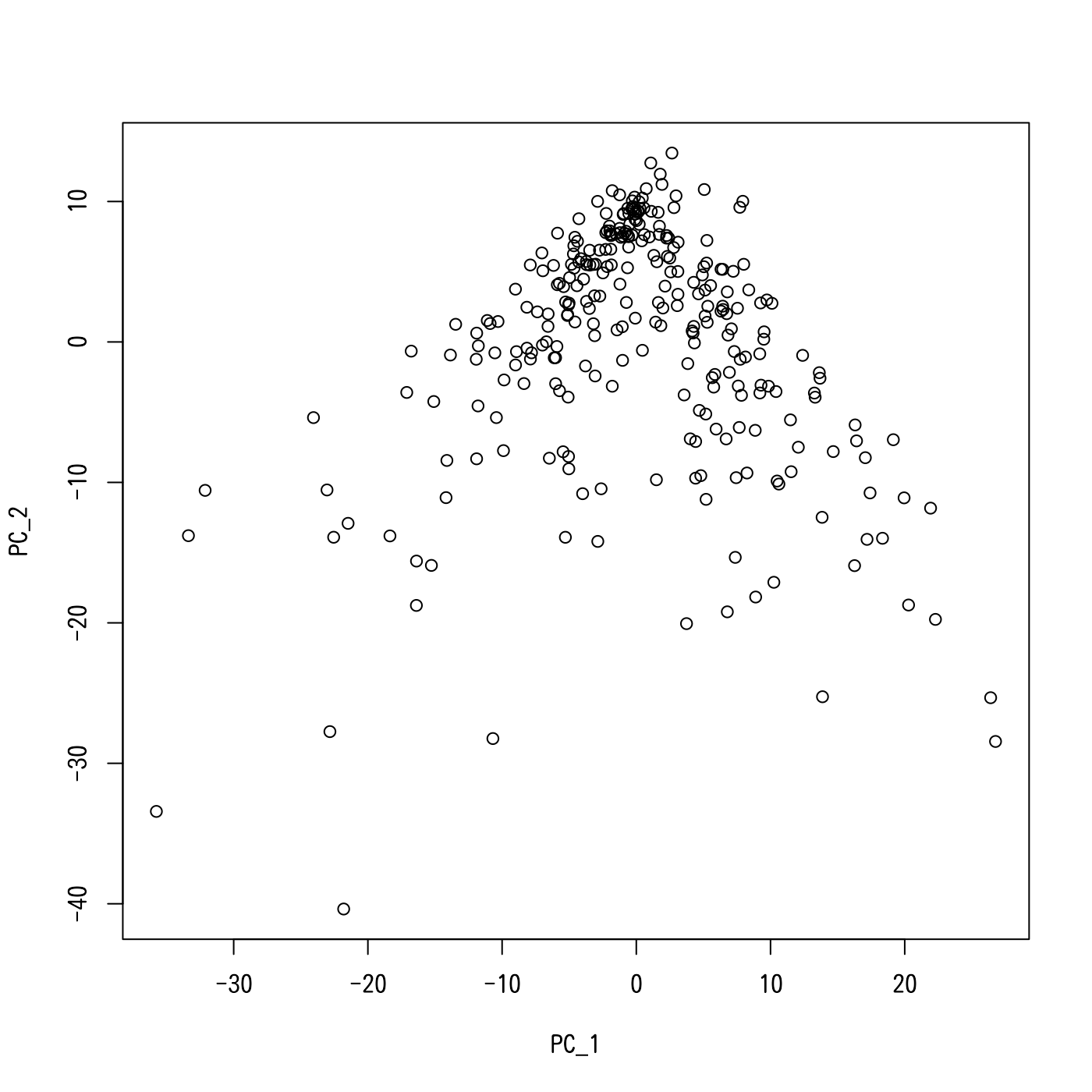
Figure 1. The result of PCA for the myoblast dataset
It is hard to reach the truth from the PCA result shown in Figure 1. Unlike the tree-like toy data analyzed in the previous tutorial (vignette("treefit")), we cannot easily recognize the underlying tree structure for various reasons (e.g., high level of noise, contamination with different types of cells, and the inaccuracy of two-dimensional visualization of high dimensional data).
Figure 2b in Trapnell el al., 2014, Nature Biotechnology shows that this dataset contains three distinct types of cells (i.e., proliferating cells, differentiating myoblasts, and contaminating interstitial mesenchymal cells), which led the authors to adopt a Y-shaped tree model (i.e., a star tree with three arms) to explain the data. However, it is difficult to quantify the reliability of this tree model and to confidently predict that the best-fit tree consists of three principal paths.
3. Quantitative trajectory inference with Treefit
3.1. Performing the Treefit analysis
Let us use Treefit in order to estimate the tree-likeness of the preprocessed myoblast data (which contain 290 cells) and to predict the number of principal paths in the best-fit tree. The workflow is the same as the toy data analysis described in the previous tutorial (vignette("treefit")).
trapnell.fit <- treefit::treefit(trapnell)
trapnell.fit## $name
## [1] "trapnell"
##
## $max_cca_distance
## p mean standard_deviation
## 1 1 0.6303459 0.12773201
## 2 2 0.4017539 0.07430735
## 3 3 0.3255355 0.05479342
## 4 4 0.2699334 0.04228203
## 5 5 0.2500198 0.04335692
## 6 6 0.2348225 0.03919659
## 7 7 0.2269746 0.03631043
## 8 8 0.2185274 0.03309986
## 9 9 0.2096228 0.03344765
## 10 10 0.2022596 0.03228294
## 11 11 0.1928535 0.03540076
## 12 12 0.1878315 0.03622790
## 13 13 0.1811885 0.03879294
## 14 14 0.1725552 0.03903983
## 15 15 0.1501188 0.03806641
## 16 16 0.1423743 0.03696181
## 17 17 0.1332141 0.03425474
## 18 18 0.1301240 0.03567304
## 19 19 0.1261766 0.03589544
## 20 20 0.1229404 0.03627929
##
## $rms_cca_distance
## p mean standard_deviation
## 1 1 0.6303459 0.12773201
## 2 2 0.5199144 0.06498940
## 3 3 0.6170262 0.06686113
## 4 4 0.6407349 0.06804858
## 5 5 0.6547144 0.05073100
## 6 6 0.5958460 0.06191394
## 7 7 0.5788654 0.05568816
## 8 8 0.6097528 0.04763631
## 9 9 0.6087060 0.05660548
## 10 10 0.6030251 0.04738148
## 11 11 0.6011495 0.04392654
## 12 12 0.6174355 0.03846905
## 13 13 0.6230418 0.03887509
## 14 14 0.6230970 0.03439415
## 15 15 0.6267405 0.03635980
## 16 16 0.6248394 0.03573201
## 17 17 0.6311507 0.02858439
## 18 18 0.6413880 0.02845259
## 19 19 0.6425895 0.03337487
## 20 20 0.6486755 0.02962193
##
## $n_principal_paths_candidates
## [1] 3 8 12 17
##
## attr(,"class")
## [1] "treefit"
plot(trapnell.fit)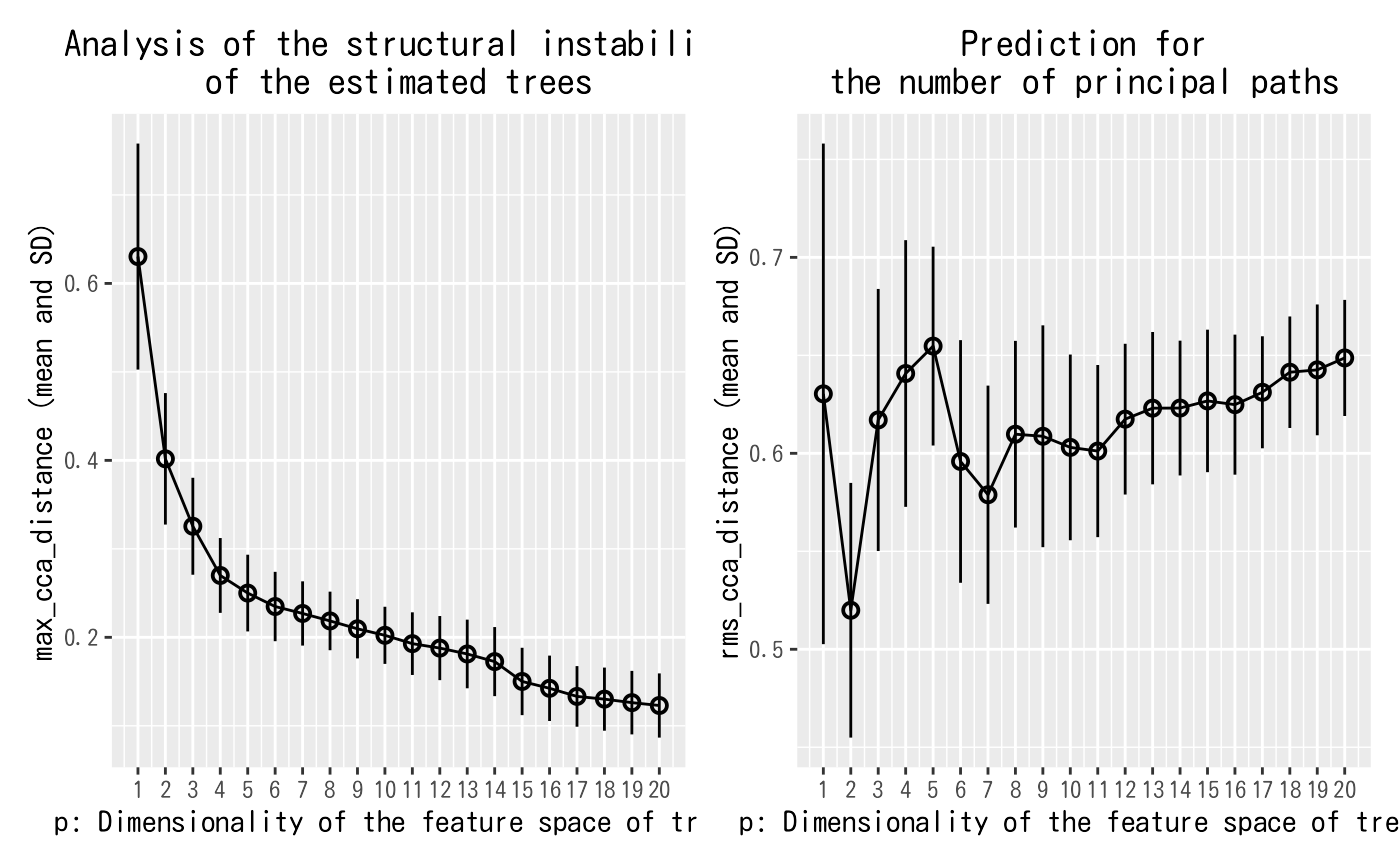
Figure 2. Summary of the Treefit analysis results
3.2. Interpreting the results
As explained in the previous tutorial (vignette("treefit")), the two Grassmann distances $max_cca_distance and $rms_cca_distance play an essential role in the Treefit analysis. The goal of the first analysis using $max_cca_distance is to measure the goodness-of-fit between data and data-derived tree trajectories, and this helps biologists make a mathematical and statistical evidence-based quantitative discussion. The other analysis with $rms_cca_distance aims to predict the number of principal paths in the best-fit tree trajectory, and the result of this analysis can be helpful to discover novel cell types or make sure whether or not there are contaminating cell types.
If we compare the mean values of $max_cca_distance shown in the left panel of Figure 2 with the previous results (vignette("treefit")), we can see that the tree-likeness of the myoblast dataset is more like the noisy tree-like data with the fatness 0.8 rather than the tree-like data with the fatness 0.1. This estimate is reasonable in light of the fact that the myoblast dataset in Trapnell el al., 2014, Nature Biotechnology contains some contaminating cell types that are not related to myogenesis.
Turning to the right panel of Figure 2, we see that $rms_cca_distance attains the local minimum at p=2. Therefore, we can deduce that the best-fit tree trajectory for the myoblast data consists of p+1=3 principal paths. $n_principal_paths_candidates[1] also tells us the number principal paths is 3. Recalling the discussion in Trapnell el al., 2014, Nature Biotechnology (i.e., the cells in the dataset can be classified into the following three groups: proliferating cells; differentiating myoblasts; and contaminating interstitial mesenchymal cells), we thus see that Treefit has correctly predicted the number of distinct principal paths forming the best-fit tree trajectory for this dataset.